AMBER
PDB fájl beszerzése és előkészítése
Az AMBER-es példához egy thrombin-binding DNA aptamert, vagyis egy egyláncú, 15 nukleotidos DNS aptamert használunk, amelyet solution NMR módszerrel határoztak meg. A szerkezet PDB kódja 1RDE. A PDB-bejegyzés szerint egy 11 konformert tartalmazó NMR ensemble-ről van szó, melynél a további példákban az első modellt használjuk.
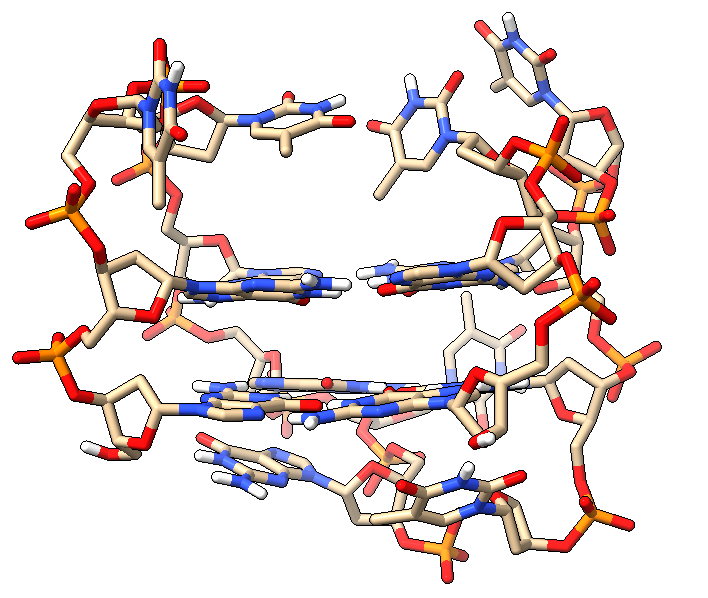
A .pdb kiterjesztésű bemeneti fájlt a PDB adatbázisából lehet beszerezni (https://www.rcsb.org/) vagy a köveztkező módon letölteni a kívánt könyvtárba.
wget https://files.rcsb.org/download/1RDE.pdb
Ez a .pdb NMR ensemble-információkat tartalmaz, illetve illetve lehetnek benne olyan rekordok és formázási elemek, amelyek nem ideálisak a LEaP/AMBER további feldolgozásához. A további számolásokhoz megfelelő formátumú .pdb-t a pdb4amber programmal lehet készíteni az alábbi módon.
pdb4amber -i 1RDE.pdb -o 1RDE_clean.pdb
Fájlok: 1RDE.pdb, 1RDE_clean.pdb
Paraméter és koordináta fájlok generálása - LEaP
A paraméter- és koordinátafájlok előállításához az AMBER beépített programja, a LEaP használható. A rendszer felépítéséhez emellett megfelelő erőteret és oldószermodellt is választani kell. Az adott AMBER-verzióhoz ajánlott erőtereket és a választható oldószermodelleket az AMBER kézikönyve tartalmazza (https://ambermd.org/Manuals.php).
Esetünkben a nukleinsavakhoz ajánlott OL21 erőteret és az OPC oldószermodellt használjuk. Mivel a vizsgált szerkezet foszfátváza negatív töltésű, a rendszer semlegesítéséhez pozitív ellenionok hozzáadása is szükséges. Az ellenionok hozzáadásával a rendszer teljes töltése semlegesíthető.
Minta tleap script (tleap_1RDE.in):
source leaprc.DNA.OL21
source leaprc.water.opc
mol = loadpdb 1RDE_clean.pdb
check mol
charge mol
addions mol Na+ 0
solvatebox mol OPCBOX 10.0
saveamberparm mol 1RDE.prmtop 1RDE.rst7
quit
Az első két sorban betöltjük a kiválasztott erőteret és az oldószermodellt. Ezután betöltjük a .pdb fájlt, és a létrehozott rendszert mol néven hivatkozzuk. A check parancs a szerkezet alapvető ellenőrzésére szolgál, míg a charge kiírja a rendszer össztöltését.
Az addions parancs a rendszer nettó töltésének semlegesítéséhez szükséges számú nátriumiont adja hozzá. A solvatebox parancs egy explicit oldószerdobozt helyez a szerkezet köré úgy, hogy a doboz fala legalább 10 Å távolságra legyen a rendszer legkülső atomjaitól.
Végül a saveamberparm parancs elmenti a számolásokhoz szükséges topológiai (.prmtop) és koordináta (.rst7) fájlokat.
A tleap script futtatása a következő módon lehetséges:
tleap -f tleap_1RDE.in
A tleap futtatása során fellépő esetleges problémák, valamint a végrehajtott lépések részletes leírása a leap.log fájlban található. Ha a tleap probléma nélkül lefutott és megvannak az 1RDE.prmtop és 1RDE.rst7 fájljaink, nekikezdhetünk a számolásoknak.
Energiaminimalizálás
A LEaP által előállított kezdeti szerkezet geometriája nem feltétlenül felel meg pontosan a választott erőtér által leírt lokális energiaminimumnak. Emellett a szolvatálás és az ionok hozzáadása után a rendszerben kedvezőtlen kontaktusok, túl közeli atom-atom távolságok vagy lokális feszültségek maradhatnak vissza. Ezért a molekuladinamikai futtatás megkezdése előtt energiaminimalizálást végzünk.
Az energiaminimalizálás célja, hogy a rendszer a legközelebbi lokális minimum irányába relaxáljon, és megszűnjenek azok a kedvezőtlen kölcsönhatások, amelyek instabilitást okozhatnának a későbbi felfűtés vagy dinamikai szimuláció során. Ez a lépés nem a globális minimum megtalálását szolgálja, hanem a kezdeti szerkezet fizikai értelemben vett “rendbetételét”.
Szolvatált rendszerek esetén célszerű a minimalizálást két lépésben elvégezni. Az első lépésben a biomolekula atomjaira pozíciós megszorításokat alkalmazunk, így elsősorban a vízmolekulák és az ionok tudnak relaxálni a szerkezet körül. A második lépésben a megszorításokat csökkentjük vagy teljesen elhagyjuk, így az egész rendszer tovább lazulhat.
Az AMBER minimalizálási bemenetében az imin=1 kapcsolja be a minimalizálást. A maxcyc a maximális ciklusszámot adja meg, míg ntmin=1 esetén az első ncyc lépés steepest descent, az azt követő lépések pedig conjugate gradient módszerrel történnek. A ntr=1 pozíciós megszorításokat kapcsol be, a restraint_wt ezek erősségét, a restraintmask pedig az érintett atomokat vagy maradékokat adja meg.
Az kényszereket tartalamzó 1_Min_rest.in:
Initial minimization with restraints
&cntrl
imin=1,
ntmin=1,
maxcyc=5000,
ncyc=2500,
ntb=1,
cut=10.0,
ntr=1,
restraint_wt=5.0,
restraintmask=':1-15',
/
Melyet lehet sander-rel és pmemd-el is futtatni a következő módon:
sander -O -i 1_Min_rest.in -o 1_Min_rest.out -p 1RDE.prmtop -c 1RDE.rst7 -r 1_Min_rest.rst7 -ref 1RDE.rst7
A -O kapcsoló lehetővé teszi a már létező kimeneti fájlok felülírását. Az -i a bemeneti fájlt adja meg, míg a -o a futás során keletkező szöveges kimeneti fájl nevét határozza meg. A -p a topológiai- vagy paraméterfájlt, a -c a kezdeti koordinátákat tartalmazó fájlt jelöli. A -r a futás végén létrejövő restart fájl nevét adja meg, amely a minimalizált koordinátákat tartalmazza.
Mivel ebben a lépésben pozíciós megszorításokat alkalmazunk, a -ref kapcsolóval meg kell adni azt a referencia-koordinátafájlt is, amelyhez képest a megszorításokat a program értelmezi.
Kimeneti fájlok: 1_Min_rest.out, 1_Min_rest.rst7
Ezt követi a kényszereket már nem tartalmazó minimalizáció (1_Min.in).
Unrestrained minimization
&cntrl
imin=1,
ntmin=1,
maxcyc=5000,
ncyc=2500,
ntb=1,
cut=10.0,
ntr=0,
/
Melynek futtatásához a korábban ismertetett parancsot használjuk a megfelelő fájlnevekkel.
sander -O -i 1_Min.in -o 1_Min.out -p 1RDE.prmtop -c 1_Min_rest.rst7 -r 1_Min.rst7
A minimalizálási lépés sikeres lefutása után a 1_Min.out fájlban ellenőrizhető a futás menete és az energia alakulása, míg a 1_Min.rst7 fájl a minimalizált koordinátákat tartalmazza.
A minimizálás akkor tekinthető sikeresnek, ha a futás hiba nélkül befejeződik, a rendszer energiája csökkenő tendenciát mutat, és az outputban nem jelennek meg rendellenes értékek, például NaN, ******** vagy extrém nagy energiájú nemkötő kölcsönhatások. Az ilyen jelenségek rendszerint rossz kontaktusokra vagy hibás kiinduló geometriára utalnak.
Amennyiben az 1_Min.out és a kapott geometria megtekintése után a minimalizációt sikeresnek könyveltük el, jöhet a következő fázis, a rendszer felmelegítése.
A rendszer felmelegítése
A minimalizálást követően a rendszert fokozatosan melegítjük fel a kívánt hőmérsékletre. A felfűtés célja, hogy a rendszer elérje a szimulációhoz szükséges hőmérsékletet anélkül, hogy hirtelen, túl nagy energiájú mozgások jelennének meg a szerkezetben. A túl gyors melegítés instabilitást okozhat, ezért a hőmérsékletet fokozatosan emeljük.
A felfűtést jellemzően állandó térfogatú (NVT) körülmények között végezzük. Ebben a lépésben a biomolekulára még célszerű enyhe pozíciós megszorításokat alkalmazni, így elsősorban az oldószer és az ionok tudnak relaxálni a szerkezet körül.
A felfűtéshez az alábbi bemeneti fájl 2_Heat.in használható:
Heating the system from 0 K to 300 K
&cntrl
imin=0,
irest=0,
ntx=1,
nstlim=25000,
dt=0.002,
ntc=2,
ntf=2,
tempi=0.0,
temp0=300.0,
ntpr=500,
ntwx=500,
ntwr=5000,
ntb=1,
ntp=0,
cut=10.0,
ntt=3,
gamma_ln=1.0,
ig=-1,
ntr=1,
restraint_wt=2.0,
restraintmask=':1-15',
nmropt=1,
/
&wt
type='TEMP0', istep1=0, istep2=20000, value1=0.0, value2=300.0,
/
&wt
type='TEMP0', istep1=20001, istep2=25000, value1=300.0, value2=300.0,
/
&wt type='END' /
A fenti minta az AMBER-es heating inputok tipikus szerkezetét követi:
imin=0→ ez már MD-futás, nem minimalizálásirest=0,ntx=1→ új futás indul koordinátákból, sebességek nélkülnstlim=25000,dt=0.002→ 50 ps futási időntb=1,ntp=0→ állandó térfogat (NVT), nyomásszabályozás nélkülntpr=500,ntwx=500,ntwr=5000→ az energetikai és futási információk kiírása 500 lépésenként történik az output fájlba, a koordináták 500 lépésenként kerülnek a trajektóriafájlba, míg a restart fájl 5000 lépésenként frissülntt=3,gamma_ln=1.0→ Langevin-termosztátnmropt=1+&wtblokkok → fokozatos hőmérséklet-rámpa 0 K-ről 300 K-re
A futtatáshoz szükséges parancs:
sander -O -i 2_Heat.in -o 2_Heat.out -p 1RDE.prmtop -c 1_Min.rst7 -r 2_Heat.rst7 -x 2_Heat.nc -ref 1_Min.rst7
A felmelegítés sikerességét a 2_Heat.out fájl alapján ellenőrizhetjük. A felmelegítés akkor tekinthető sikeresnek, ha a rendszer hőmérséklete fokozatosan közelíti a beállított célértéket, a futás hiba nélkül befejeződik és az outputban nem jelennek meg rendellenes értékek, például NaN vagy ********. Továbbá érdemes vizuálisan megtekinteni a kapott trajektóriát (2_Heat.nc), hogy nincsenek e irreálisan nagy eltorzulások a nukleinsav esetén, illetve a vizes doboz is megfelelően viselkedik e. A futás végén létrejövő 2_Heat.rst7 fájl a következő, kiegyenlítési szakasz bemeneteként használható.
Kiegyenlítés
A felfűtést követően a rendszert állandó nyomású körülmények között kiegyenlítjük. Ennek célja, hogy a vízdoboz mérete és sűrűsége, valamint az oldószer és az ionok elrendeződése alkalmazkodjon a nukleinsav-szerkezethez.
Explicit oldószeres AMBER-szimulációk esetén a kiegyenlítési szakasz tipikusan a heating végén kapott koordinátákból és sebességekből indul, ezért restart futásként történik. A rendszer ebben a lépésben már a kívánt hőmérsékleten marad, miközben a nyomásszabályozás lehetővé teszi, hogy a szimulációs doboz mérete a megfelelő sűrűséghez igazodjon.
A kiegyenlítéshez az alábbi 3_Equil.in fájl használható:
Equilibration at 300 K and 1 atm without restraints
&cntrl
imin=0,
irest=1,
ntx=5,
nstlim=50000,
dt=0.002,
ntc=2,
ntf=2,
temp0=300.0,
ntpr=500,
ntwx=500,
ntwr=5000,
ntb=2,
ntp=1,
taup=2.0,
cut=10.0,
ntt=3,
gamma_ln=1.0,
ig=-1,
ntr=0,
/
A fenti kiegyenlítési futás AMBER-es szempontból tipikus beállításokat használ:
imin=0→ molekuladinamikai futás, nem energiaminimalizálásirest=1,ntx=5→ restartból induló futás, a felfűtés végén kapott koordináták és sebességek felhasználásávalnstlim=50000,dt=0.002→ 100 ps szimulációs időntb=2,ntp=1→ állandó nyomású (NPT) futás, amely lehetővé teszi a dobozméret változásáttaup=2.0→ a nyomásszabályozás relaxációs idejetemp0=300.0→ célhőmérséklet 300 Kntt=3,gamma_ln=1.0→ Langevin-termosztát a hőmérséklet szabályozásárantc=2,ntf=2→ a hidrogént tartalmazó kötések korlátozása, amely lehetővé teszi a 2 fs időlépéstntr=0→ ebben a szakaszban már nem alkalmazunk pozíciós megszorításokatntpr=500,ntwx=500,ntwr=5000→ az energetikai és futási információk 500 lépésenként íródnak az output fájlba, a koordináták 500 lépésenként kerülnek a trajektóriafájlba, míg a restart fájl 5000 lépésenként frissül
A futtatáshoz szükséges parancs:
sander -O -i 3_Equil.in -o 3_Equil.out -p 1RDE.prmtop -c 2_Heat.rst7 -r 3_Equil.rst7 -x 3_Equil.nc
Kimeneti fájlok: 3_Equil.out, 3_Equil.nc, 3_Equil.rst7
A kiegyenlítési szakasz végén ellenőrizni kell, hogy a futás hiba nélkül befejeződött-e, valamint létrejöttek-e a megfelelő kimeneti fájlok. A sűrűség, a hőmérséklet és a dobozméret viselkedése alapján megítélhető, hogy a rendszer közelít-e az egyensúlyi állapothoz. Mivel ebben a példában OPC oldószermodellt használunk, amelyet kifejezetten a folyékony víz tömbi tulajdonságainak - köztük a sűrűségnek - jó reprodukálására fejlesztettek ki, a beállt sűrűségnek egy szolvatált biomolekulás rendszerben is a folyékony víz sűrűségéhez közeli, nagyjából 1.0 g/cm^3 körüli tartományban kell lennie; a biomolekula és az ionok jelenléte miatt kisebb eltérés természetes lehet. Emellett fontos, hogy az outputban ne jelenjen meg NaN vagy ********, és a trajektória vizuálisan is ésszerű szerkezeti viselkedést mutasson. Ha ezek a feltételek teljesülnek, a rendszer alkalmas a produkciós futás megkezdésére.
Mintavételezési szakasz
A kiegyenlítési szakasz sikeres lefutása után a következő lépés a mintavételezési szakasz elindítása. Ebben a fázisban a rendszer már a kívánt hőmérsékleten és nyomáson, megszorítások nélkül fejlődik tovább, így a kapott trajektória alkalmas a szerkezeti és dinamikai tulajdonságok kiértékelésére. A mintavételezéses futás a kiegyenlítés végén kapott koordinátákból és sebességekből indul, ezért restart futásként történik.
Egy 20 ns hosszúságú, minta 4_MD.in fájl:
Production MD at 300 K and 1 atm
&cntrl
imin=0,
irest=1,
ntx=5,
nstlim=12500000,
dt=0.002,
ntc=2,
ntf=2,
temp0=300.0,
ntpr=10000,
ntwx=10000,
ntwr=500000,
ioutfm=1,
ntb=2,
pres0=1.0,
ntp=1,
taup=2.0,
cut=10.0,
ntt=3,
gamma_ln=1.0,
ig=-1,
ntr=0,
/
A fenti produkciós futás AMBER-es szempontból tipikus beállításokat használ:
imin=0→ molekuladinamikai futás, nem energiaminimalizálásirest=1,ntx=5→ restartból induló futás, a korábbi sebességek és koordináták felhasználásávalnstlim=12500000,dt=0.002→ 25 ns szimulációs időntb=2,ntp=1→ állandó nyomású (NPT) futástemp0=300.0→ célhőmérséklet 300 Kntt=3,gamma_ln=1.0→ Langevin-termosztátntr=0→ nincs pozíciós megszorításioutfm=1→ NetCDF formátumú trajektória
A 4_MD.in futtatásához szüksége sparancs:
pmemd -O -i 4_MD.in -o 4_MD.out -p 1RDE.prmtop -c 3_Equil.rst7 -r 4_MD.rst7 -x 4_MD.nc -inf 4_MD.mdinfo
Note
A futtatáshoz használt programot érdemes az adott hardverkörnyezethez igazítani. Általános, CPU-alapú futtatásra a sander vagy a teljesítményre optimalizált pmemd használható, míg GPU-s rendszeren a pmemd.cuda javasolt, mivel ez az AMBER GPU-ra optimalizált molekuladinamikai motorja. A több CPU-maggal történő párhuzamos futtatás MPI-s környezetben tipikusan pmemd.MPI vagy sander.MPI használatával történik.
A futás során több fontos kimeneti fájl jön létre:
Analízis: trajektória RMSD-je és hidrogénkötések számának meghatározása cpptraj-al
Első analízisként a trajektória RMSD-je számítható ki a cpptraj programmal. A számolás előtt célszerű a trajektóriát autoimage paranccsal újraimázsolni, hogy a periodikus határfeltételek miatt szétszórtnak látszó rendszer újra értelmezhetően jelenjen meg. Az RMSD ezután számolható az első frame-hez vagy egy külön referencia-szerkezethez viszonyítva. A trajout paranccsal egy új, vizuálisan „szép” trajektória is elmenthető.
A következő minta csak a nukleinsav nem hidrogén atomjainak RMSD-jét határozza meg (cpptraj_rmsd.in):
parm 1RDE.prmtop
trajin 4_MD.nc
autoimage
rms first :1-15&!@H= out rmsd_1RDE.dat
trajout 4_MD_autoimaged.nc netcdf
run
quit
Az RMSD vizualizációját pedig a következő kis python scripttel lehet megtenni (rmsd_plot.py):
import matplotlib.pyplot as plt
frames = []
rmsd = []
with open("rmsd_1RDE.dat") as f:
for line in f:
if line.strip() and not line.startswith("#"):
parts = line.split()
frames.append(int(parts[0]))
rmsd.append(float(parts[1]))
# Plot készítése
plt.figure(figsize=(8,5))
plt.plot(frames, rmsd, color='blue', linewidth=2)
plt.xlabel("Frame")
plt.ylabel("RMSD [Å]")
plt.title("TBA tetrad RMSD during MD simulation")
plt.grid(True)
plt.tight_layout()
plt.savefig("rmsd_tetrad_plot.png", dpi=300) # elmentjük képként
plt.show()
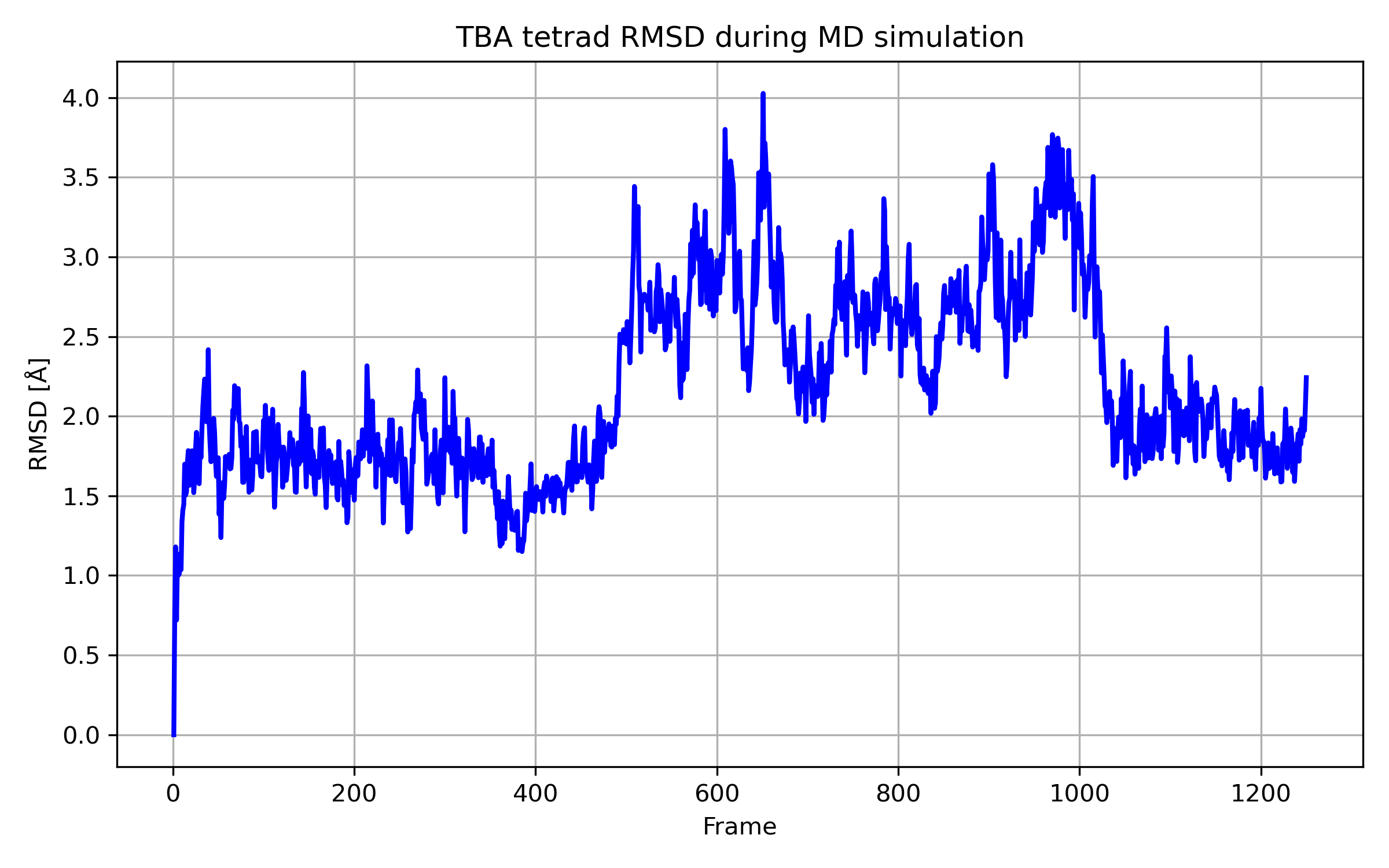
A trajektóriafájl vizualizációja után a kapott RMSD görbe logikus képet ad, hiszen a szerezet a dinamikai szimuláció során elkezdett szétesni, majd az utolsó 5 ns alatt újra visszatért a kiindulási szerkezethez hasonló geometriába, melynél a tetrádokat másodrendű hidrogénkötések stabilizálják.
A 2 guanin tetrádban jelenlévő hidrogénkötések számát cpptraj-al szintén ki lehet irattatni a következő módon (cpptraj_hbonds.in):
parm 1RDE.prmtop
trajin 4_MD_autoimaged.nc
# Hoogsteen H-bonds between G-tetrad guanines only
hbond Hoogsteen_hbonds \
donormask :1-2,5-6,10-11,14-15 \
acceptormask :1-2,5-6,10-11,14-15@N7,O6 \
dist 3 \
angle 150 \
out hbond_hoogsteen.dat \
avgout hbond_hoogsteen_avg.dat
run
Ez a geometria alapján a 3Å távolságon belül és legfeljebb 150°-os szöget bezáró donor-akceptor egységeket számolja, mint hidrogénkötéseket. A korábbihoz hasonló python scripttel (hbonds_tetrad_plot.py) pedig ki lehet rajzoltatni a hidrogénkötések szánának framenkénti változását a hbond_hoogsteen.dat fájl segítségével.
import matplotlib.pyplot as plt
hbonds = []
with open("hbond_hoogsteen.dat") as f:
for line in f:
if line.startswith("#"):
continue
parts = line.split()
if len(parts) >= 2:
frames.append(int(parts[0]))
hbonds.append(int(parts[1]))
plt.figure(figsize=(8,5))
plt.plot(frames, hbonds, linestyle='-', color='r')
plt.xlabel("Frame")
plt.ylabel("Number of Hoogsteen H-bonds")
plt.title("Evolution of TBA tetrad H-bonds during the simulation")
plt.grid(True)
plt.tight_layout()
plt.savefig("hbonds_tetrad_plot.png", dpi=300)
plt.show()
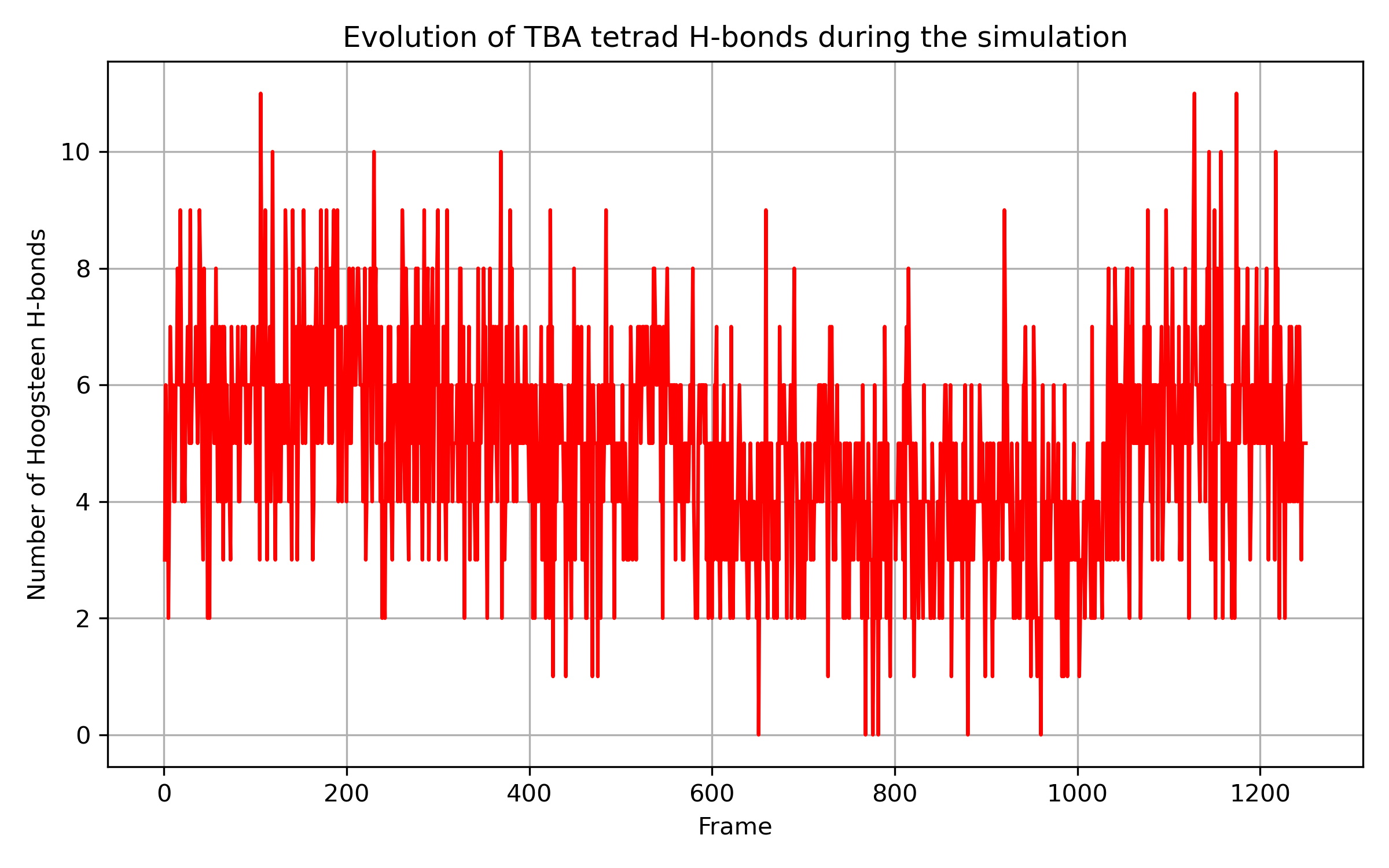
A kapott eredmények szépen mutatják, hogy a hidrogénkötések száma átlagosan 2-vel csökkent, mikor a szerkezet kissé szétnyílt (400-1000 framek).