GROMACS
A GROMACS egy széles körben használt, nyílt forráskódú molekuladinamikai programcsomag, amely biomolekulák, oldatok és más molekuláris rendszerek szimulációjára és analízisére szolgál. A program különösen elterjedt fehérjék, nukleinsavak, membránrendszerek és kisebb molekulák molekuladinamikai vizsgálatában.
A GROMACS a szimulációs munkafolyamat teljes láncát támogatja: a bemeneti struktúra előkészítésétől kezdve a topológia létrehozásán, a rendszer dobozolásán, szolvatálásán és ionizálásán át az energiaminimalizálásig, a kiegyenlítésig, a produkciós futásig és az utólagos analízisig.
A következő fejezetekben egy tipikus GROMACS-workflow lépéseit mutatjuk be egy fehérje vízben történő szimulációjának példáján keresztül.
Tesztrendszer
A GROMACS használatának bemutatásához egy klasszikus, széles körben alkalmazott példarendszert választunk: a lizozim vizes közegben történő szimulációját. Ez a rendszer különösen jól alkalmas a molekuladinamikai szimulációk alapvető lépéseinek bemutatására, mivel egyetlen workflow-ban szemléltethető a topológia létrehozása, a dobozolás, a szolvatálás, az ionok hozzáadása, az energiaminimalizálás, a kiegyenlítés és a mintavételezési szakasz.
A bemeneti szerkezet előkészítése
A GROMACS-os szimuláció előkészítésének első lépése a kiindulási szerkezet ellenőrzése és megtisztítása. A példában egy lizozim PDB-fájljából indulunk ki, amelyet a további lépésekhez GROMACS-kompatibilis formára kell előkészíteni. A klasszikus lysozyme in water tutorial is azzal kezdődik, hogy a felhasználó letölti a megfelelő PDB-fájlt, vizualizálja a szerkezetet, majd eltávolítja a nem szükséges elemeket, például a kristályvizeket.
A .pdb kiterjesztésű bemeneti fájlt (1AKI.pdb) a PDB adatbázisából lehet beszerezni (https://www.rcsb.org/) vagy a köveztkező módon letölteni a kívánt könyvtárba.
wget https://files.rcsb.org/download/1AKI.pdb
A kiindulási PDB-fájl ellenőrzésénél különösen fontos az alábbiakra figyelni:
a szerkezet tartalmaz-e kristályvizeket (például
HOHmaradékokat), amelyeket a legtöbb alapvető vizes szimulációs workflow-ban eltávolítunk,van-e a fájlban olyan ligand, kofaktor vagy egyéb
HETATMrekord, amelyre az adott példában nincs szükség,szerepel-e a PDB-megjegyzések között hiányzó atomra vagy hiányzó maradékra utaló információ, mert ezek a későbbi
pdb2gmxfutását megakaszthatják,a rendszer valóban csak azt a makromolekulát tartalmazza-e, amelyet szimulálni szeretnénk.
A PDB-fájl vizuális ellenőrzése ajánlott még a GROMACS-lépések előtt. Erre bármely általánosan használt molekulamegjelenítő program használható. A lysozyme-os tutorialok is javasolják, hogy a szerkezetet először nézzük meg, mielőtt a topológiaépítéshez továbbhaladunk.
A kristályvizek eltávolítása legegyszerűbben szövegszerkesztővel vagy parancssorból történhet. A GROMACS-os példák tipikusan abból indulnak ki, hogy a bemeneti PDB-fájl csak a szükséges fehérjeatomokat tartalmazza. A kristályvizek eltávolítása azért szokásos, mert a rendszert később úgyis újra szolvatáljuk egy definiált vízmodellel. Egy tipikus parancssori tisztítási példa:
grep -v HOH 1AKI.pdb > 1AKI_clean.pdb
Ez a parancs eltávolítja a HOH jelölésű sorokat a fájlból, és egy új, tisztított PDB-fájlt hoz létre (1AKI_clean.pdb). Hasonló módon más, nem kívánt rekordok is szűrhetők.
A bemeneti szerkezet előkészítésének célja tehát az, hogy a következő lépésben a pdb2gmx egy tiszta, konzisztens, hiányzó elemeket nem tartalmazó fehérjeszerkezeten dolgozhasson. Ez alapvető feltétele a topológia sikeres létrehozásának.
Topológia létrehozása
A megtisztított PDB-fájl előkészítése után a következő lépés a fehérje topológiájának létrehozása. GROMACS-ban ezt tipikusan a gmx pdb2gmx paranccsal végezzük el, amely a bemeneti fehérjeszerkezetből GROMACS-kompatibilis koordinátafájlt, topológiai fájlt és szükség esetén pozíciós restrikciós fájlokat generál.
A példában a topológia létrehozásához a következő parancsot használtuk:
gmx pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water tip3p
Ebben a parancsban a -f kapcsoló adja meg a bemeneti PDB-fájlt, a -o az elkészítendő GROMACS-formátumú koordinátafájlt, a -water tip3p pedig a választott vízmodellt. A pdb2gmx futtatása során a program megkérdezi, hogy melyik force fieldet szeretnénk használni; ebben a példában a CHARMM27 parametrizációt választottuk.
A GROMACS dokumentáció szerint a CHARMM27 egy hivatalosan támogatott, all-atom CHARMM force field GROMACS-ban. A force field kiválasztása alapvetően meghatározza a topológiai paramétereket, az atomtípusokat, a töltéseket és a kötési kölcsönhatásokat, ezért fontos, hogy a későbbi szimulációs beállítások is összhangban legyenek ezzel a választással.
A pdb2gmx futtatása után tipikusan az alábbi fontos fájlok jönnek létre:
1AKI_processed.gro→ a feldolgozott, GROMACS-formátumú koordinátafájl,topol.top→ a rendszer fő topológiai fájlja,posre.itp→ a fehérjére vonatkozó pozíciós restrikciós fájl, amelyet jellemzően a kiegyenlítési lépésekben használunk.
A topológia létrehozása után érdemes ellenőrizni, hogy a feldolgozott szerkezet vizuálisan is ésszerűnek tűnik-e.
A rendszer dobozolása és szolvatálása
A topológia létrehozása után a fehérjét egy megfelelő méretű szimulációs dobozba kell helyezni, majd a dobozt oldószerrel kell feltölteni. A GROMACS-os fehérje-víz workflow-ban ez a lépés tipikusan a gmx editconf és a gmx solvate parancsokkal történik.
A dobozolás során az a cél, hogy a fehérje és a doboz szélei között elegendő távolság legyen, így elkerülhetők a periodikus határfeltételekből származó mesterséges kölcsönhatások. A GROMACS-os példákban ezért jellemzően meghatározunk egy minimális távolságot a makromolekula és a doboz határa között.
A példában a dobozoláshoz a következő parancsot használtuk:
gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.2 -bt cubic
Ebben a parancsban a -f kapcsoló adja meg a bemeneti koordinátafájlt, a -o az új koordinátafájl nevét, a -c a fehérje középre helyezését kéri, a -d 1.2 pedig azt írja elő, hogy a fehérje és a doboz széle között legalább 1.2 nm távolság legyen. A -bt cubic a doboz alakját köbösre állítja.
A dobozolás után a rendszert vízmolekulákkal kell feltölteni. A példában ehhez a következő parancsot használtuk:
gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
Ebben a parancsban a -cp a dobozolt fehérjét tartalmazó koordinátafájlt adja meg, a -cs a felhasznált oldószerkonfigurációt,
az -o a szolvatált rendszer új koordinátafájlját, a -p topol.top pedig azt biztosítja, hogy a topológiai fájl automatikusan frissüljön a hozzáadott oldószermolekulák számával.
A dobozolás és a szolvatálás után érdemes ellenőrizni, hogy
létrejött-e a
1AKI_newbox.grofájl, amely már tartalmazza a dobozméretet és a középre helyezett fehérjét,létrejött-e a
1AKI_solv.grofájl, amely a vízzel feltöltött rendszert tartalmazza,a
topol.topfájl frissült-e az oldószermolekulák számával,a fehérje és a doboz széle közötti távolság megfelelőnek tűnik-e, és a szolvatált rendszer vizuálisan is ésszerű-e.
Ionok hozzáadása
A szolvatált rendszer létrehozása után a következő lépés az ionok hozzáadása. Ennek elsődleges célja a rendszer nettó töltésének semlegesítése, valamint szükség esetén egy adott sókoncentráció beállítása.
Az ionok hozzáadásához először egy .tpr futtatási bemenetet kell generálni a gmx grompp segítségével. Ehhez általában egy egyszerű, energiamentesítési jellegű ions.mdp fájlt használunk, mert a genion programnak szüksége van egy előkészített rendszerleírásra.
Egy általános ions.mdp fájl:
; ions.mdp - used as input into grompp to generate ions.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Minimization step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; Method to determine neighbor list (simple, grid)
coulombtype = cutoff ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
Egy tipikus előkészítő parancs a következő:
gmx grompp -f ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
Ebben a parancsban a -f kapcsoló az ions.mdp paraméterfájlt adja meg, a -c a szolvatált koordinátafájlt, a -p a topológiai fájlt, az -o pedig az elkészítendő ions.tpr bináris futtatási bemenetet.
Ezután az ionok hozzáadása a gmx genion paranccsal történik. Egy egyszerű, semlegesítést végző példa:
gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
Ebben a parancsban a -s kapcsoló a grompp által létrehozott ions.tpr fájlt adja meg, az -o a kimeneti koordinátafájlt, a
-p topol.top pedig biztosítja, hogy a topológiai fájl frissüljön az ionok számával. A -pname NA és -nname CL opciók a pozitív és negatív ionok nevét állítják be, a -neutral pedig azt kéri, hogy a program a rendszer nettó töltését semlegesítse.
A parancs futtatásakor a program rákérdez, hogy melyik atomcsoport molekuláit cserélje le ionokra. Ebben a példában a 13 számú
SOL csoportot kell kiválasztani, vagyis a vízmolekulákat, mert az ionokat az oldószer helyére szeretnénk beilleszteni, nem pedig a fehérje atomjait helyettesíteni.
Ha nemcsak semlegesíteni szeretnénk a rendszert, hanem például fiziológiás sókoncentrációt is be szeretnénk állítani, akkor a genion parancsban a -conc opció is használható. Például a -conc 0.150 hozzávetőlegesen 150 mM sókoncentráció beállítását kéri, a -neutral opcióval együtt pedig a rendszer nettó töltése is semleges marad. Ez a megoldás több fehérje-víz tutorialban is szerepel.
Az ionozás után érdemes ellenőrizni, hogy létrejött-e a 1AKI_solv_ions.gro fájl, amely már tartalmazza a hozzáadott ionokat.
Az ionok hozzáadásával elkészül az a szolvatált és semlegesített rendszer, amely már közvetlenül felhasználható az energiaminimalizálási lépéshez.
Energiaminimalizálás
Az energiaminimalizálás célja, hogy a rendszerből eltávolítsuk a kedvezőtlen közeli kontaktusokat, a szterikus ütközéseket és az esetleges rossz lokális geometriát, mielőtt a tényleges molekuladinamikai futások megkezdődnének.
A GROMACS dokumentáció szerint az energiaminimalizálás több algoritmussal is végezhető, de biomolekulás rendszerek előkészítéséhez a
steepest descent eljárás az egyik leggyakoribb választás, mert robusztus és egyszerűen alkalmazható. Az algoritmus addig fut, amíg vagy el nem ér egy felhasználó által meghatározott erőküszöböt, vagy el nem fogy az engedélyezett lépésszám.
Az energiaminimalizálás futtatásához szükség van egy minim.mdp fájlra is, amely a minimizálás paramétereit tartalmazza. Ebben adjuk meg többek között a használt algoritmust (például steep), a konvergenciaküszöböt (emtol), a lépésméretet (emstep) és a maximális lépésszámot (nsteps), vagyis ez a fájl szabja meg, hogyan végezze el a GROMACS az energiaminimalizálást.
Minta minim.mdp fájl:
; minim.mdp - used as input into grompp to generate em.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Minimization step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; Method to determine neighbor list (simple, grid)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
Ebben a példában az integrator = steep a steepest descent algoritmust választja, az emtol = 1000.0 a célként kitűzött
maximális erőküszöböt adja meg kJ mol-1nm-1 egységben, az emstep = 0.01 a maximális lépésméretet, az
nsteps = 50000 pedig a megengedett maximális lépésszámot. A nemkötő kölcsönhatások és a peremfeltételek beállításai szintén ebben
a fájlban szerepelnek.
Az energiaminimalizálás futtatásához először egy .tpr bemenetet kell készíteni a gmx grompp paranccsal.
gmx grompp -f minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
Ebben a parancsban a -f kapcsoló az energiaminimalizálási paraméterfájlt adja meg, a -c a kiindulási koordinátafájlt, a -p a topológiai fájlt, az -o pedig az elkészítendő em.tpr bináris futtatási bemenetet. A grompp feladata, hogy a koordinátákat, a topológiát és az .mdp beállításokat egyetlen futtatási bemenetté egyesítse.
A minimalizálás tényleges futtatása ezután a gmx mdrun paranccsal történik:
gmx mdrun -v -deffnm em
A -v opció részletesebb futáskövetést ad a képernyőre, a -deffnm em pedig azt jelenti, hogy a program az em alapnévvel
hozza létre a futáshoz tartozó bemeneti és kimeneti fájlokat. A GROMACS dokumentáció szerint a mdrun nemcsak molekuladinamikai szimulációra, hanem energiaminimalizálásra is használható, ha a .tpr fájl ezt a feladatot írja elő.
A futás eredményeként tipikusan létrejönnek az alábbi fájlok:
em.tpr→ az energiaminimalizálás bináris futtatási bemenete,em.log→ a futás naplófájlja, amely a konvergenciára és az erőkre vonatkozó információkat is tartalmazza,em.edr→ az energiafájl, amelyből később az energiakomponensek kiolvashatók,em.gro→ az energiaminimalizált szerkezet, amely a későbbi kiegyenlítési lépések kiindulási geometriája lesz.
Sikeres futás esetén a terminálban vagy az em.log fájl végén a következőhöz hasonló üzenet jelenik meg:
Steepest Descents converged to Fmax < 1000 in ... steps
Ez azt jelzi, hogy a steepest descent algoritmus elérte a kívánt erőküszöböt. A lysozyme tutorial ezt tekinti az egyik legfontosabb jelének annak, hogy az energiaminimalizálás megfelelően lefutott.
A következő paranccsal kiírattatható a potenciális energia változása egy potential.xvg fájlba:
gmx energy -f em.edr -o potential.xvg
A gmx energy futtatásakor a promptnál a 11 0 értékeket kell megadni: a 11 a Potential energiatag kiválasztását jelenti, a 0 pedig a bevitelt zárja le.
Melyet akár egy rövid python script (potential_plot.py) segítségével ki is lehet rajzoltatni.
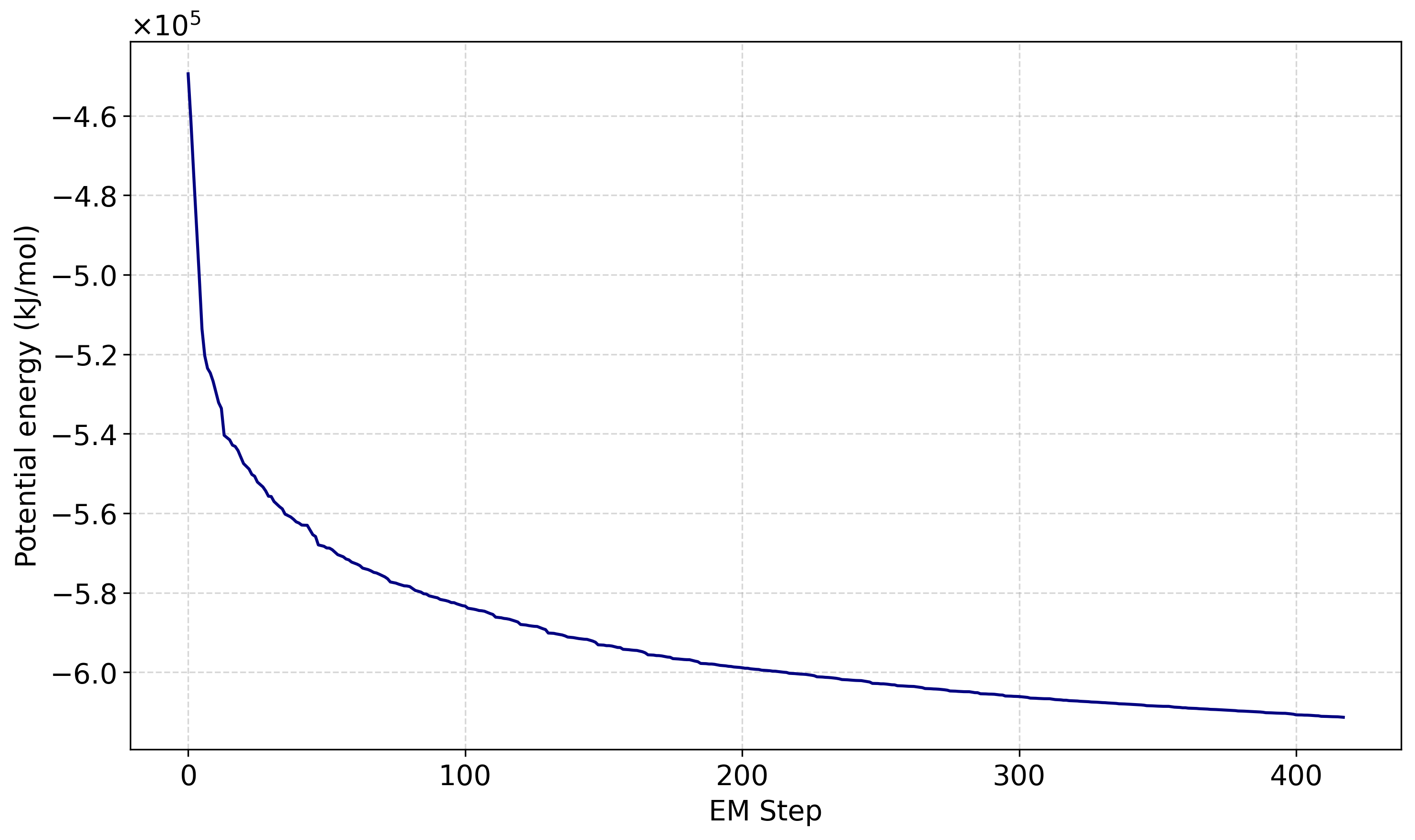
Az energiaminimalizálással előállított em.gro szerkezet lesz a következő lépés, vagyis az NVT kiegyenlítés kiindulási geometriája.
NVT kiegyenlítés
Az energiaminimalizálás után a rendszer még nem tekinthető teljesen kiegyenlítettnek. Bár a geometriai feszültségek és a szterikus ütközések ekkorra már nagyrészt megszűntek, a vízmolekuláknak és az ionoknak még alkalmazkodniuk kell a fehérje környezetéhez. A kiegyenlítés során a pdb2gmx által korábban létrehozott posre.itp fájlt használjuk. A GROMACS dokumentáció szerint a pozíciós restrikciók célja ilyenkor az, hogy a kritikus részeket — például a fehérje nehézatomjait — a helyükön tartsuk, miközben az oldószer és az ionok rendeződnek körülöttük.
Az NVT szakasz célja a rendszer hőmérsékletének stabilizálása állandó térfogat mellett. A lysozyme tutorial szerint ez a fázis jellemzően rövidebb, és általában elegendő arra, hogy a rendszer elérje a kívánt hőmérsékletet. A példában az NVT lépés az energiaminimalizált szerkezetből indul.
Az NVT-hez szükség van egy nvt.mdp fájlra is, amely a hőmérsékleti kiegyenlítés paramétereit tartalmazza. A lysozyme tutorialban külön kiemelik, hogy ebben a szakaszban a fontosabb beállítások közé tartozik a gen_vel = yes, amely kezdeti sebességeket generál, a tcoupl = V-rescale, amely a hőmérséklet-szabályozást végzi, valaminta pcoupl = no, mert ebben a szakaszban még nincs nyomáscsatolás.
Minta nvt.mdp fájl:
title = OPLS Lysozyme NVT equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 2500 ; save coordinates every 5.0 ps
nstvout = 2500 ; save velocities every 5.0 ps
nstenergy = 2500 ; save energies every 5.0 ps
nstlog = 2500 ; update log file every 5.0 ps
; Bond parameters
continuation = no ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
; vdW
rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
rvdw-switch = 1.0
vdw-modifier = force-switch
DispCorr = No ; per CHARMM FF convention
; Electrostatics
rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm)
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; stochastic Bussi thermostat
tc-grps = System
tau_t = 1.0 ; value of tau (ps)
ref_t = 298 ; temperature (K)
; Pressure coupling is off
pcoupl = no ; no pressure coupling in NVT
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 298 ; temperature for Maxwell distribution
gen_seed = -1 ; generate a random seed
Az NVT futtatásához először egy nvt.tpr futtatási bemenetet kell készíteni:
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
Ebben a parancsban a -f kapcsoló az NVT paraméterfájlt adja meg, a -c em.gro az energiaminimalizált szerkezetet, a -r em.gro pedig a pozíciós restrikciók referencia-koordinátáit. A lysozyme tutorial kifejezetten hangsúlyozza, hogy a restrainelt equilibration során a -r opcióval meg kell adni azt a koordinátafájlt, amelyhez a restraintek viszonyulnak.
Az NVT futás ezután a következő paranccsal indítható:
gmx mdrun -deffnm nvt
A -deffnm nvt opció azt jelenti, hogy a GROMACS a futás során nvt előtaggal hozza létre a bemeneti és kimeneti fájlokat, például nvt.log, nvt.edr, nvt.cpt és nvt.gro formában. A mdrun a GROMACS általános futtatómotorja, amely a .tpr fájlban megadott feladatnak megfelelően hajtja végre a szimulációt. Az NVT futás után érdemes ellenőrizni, hogy a rendszer hőmérséklete megfelelően stabilizálódott-e.
A következő paranccsal kiírattatható a hőmérséklet változása egy temperature.xvg fájlba:
gmx energy -f nvt.edr -o temperature.xvg
A gmx energy futtatásakor a promptnál a 16 0 értékeket kell megadni: a 16 a rendszer Temperature tagjának kiválasztását jelenti, a 0 pedig a bevitelt zárja le.
Melyet később akár egy rövid python script (temperature_plot.py) segítségével ki is lehet rajzoltatni.
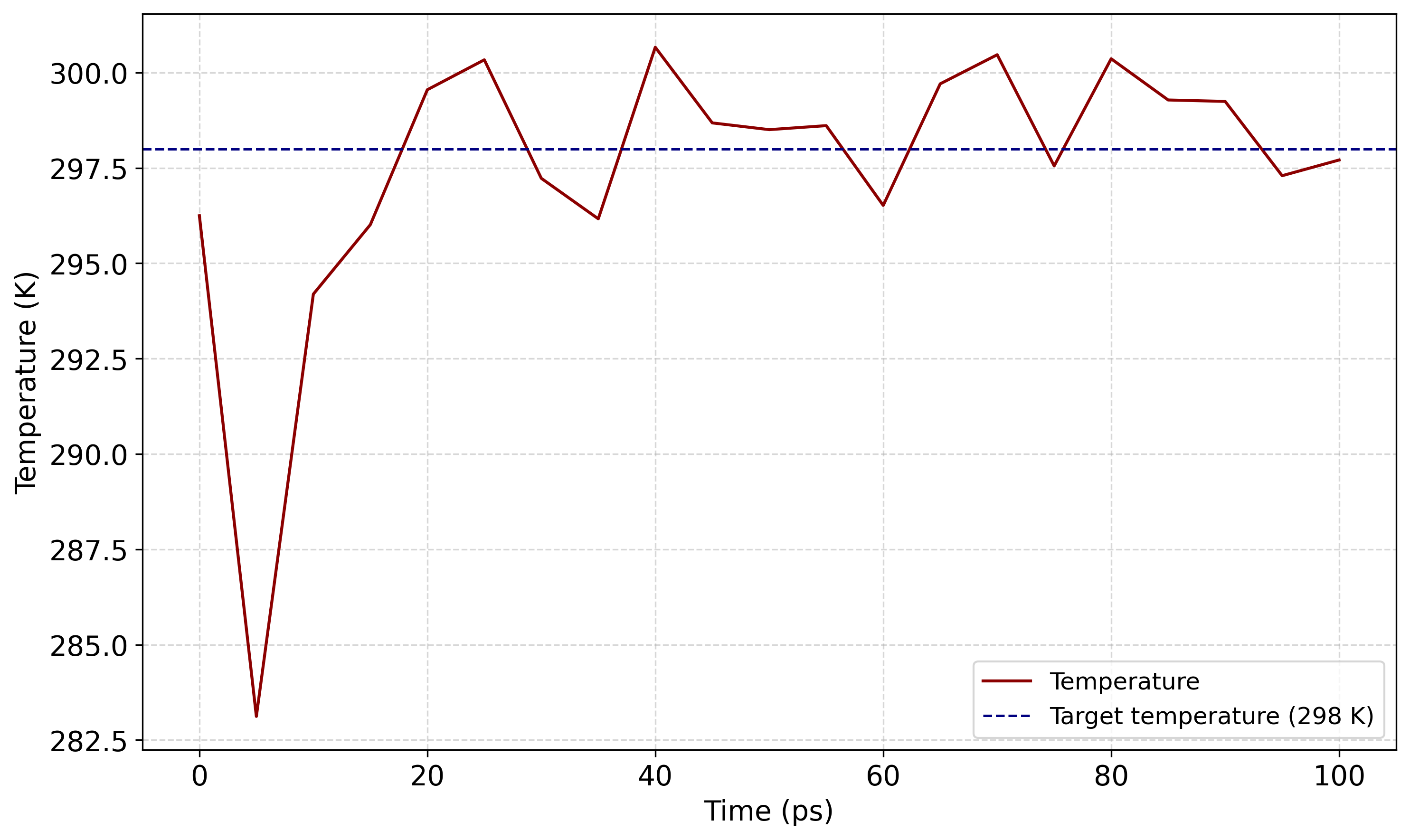
NPT kiegyenlítés
A következő lépés az NPT kiegyenlítés, amelynek célja a nyomás és ezzel együtt a sűrűség stabilizálása.
Az NPT futtatásához szükség van egy npt.mdp fájlra is, amely az NVT paramétereihez képest már nyomáscsatolási beállításokat is tartalmaz. Ebben a szakaszban fontos, hogy a futást az NVT-ből folytassuk, ezért a tipikus beállítások között szerepel a continuation = yes és a gen_vel = no opció. Ebben a fázisban már nem generálunk új sebességeket, hanem azokat az előző szakaszból vesszük át.
Minta npt.mdp fájl:
title = OPLS Lysozyme NPT equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 250000 ; 2 * 250000 = 500 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 500 ; save coordinates every 1.0 ps
nstvout = 500 ; save velocities every 1.0 ps
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
; Bond parameters
continuation = yes ; Restarting after NVT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
; vdW
rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
rvdw-switch = 1.0
vdw-modifier = force-switch
DispCorr = No
; Electrostatics
rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm)
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; stochastic Bussi thermostat
tc-grps = System
tau_t = 1.0
ref_t = 298
; Pressure coupling is on
pcoupl = C-rescale
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 5.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
refcoord_scaling = com
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = no ; Velocity generation is off
Az NPT futtatási bemenetet (npt.tpr) a következő paranccsal készíthetjük el:
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
Ebben a parancsban a -c nvt.gro az NVT szakasz végső koordinátáit adja meg, a -r nvt.gro továbbra is a pozíciós restrikciók referenciaállapota, míg a -t nvt.cpt biztosítja, hogy a rendszer állapota folytonosan folytatódjon az NVT szakaszból.
Az NPT futás indítása:
gmx mdrun -deffnm npt
A futás eredményeként tipikusan létrejönnek az npt.gro, npt.edr, npt.log és npt.cpt fájlok. Ezek közül az npt.gro lesz a produkciós szimuláció kiindulási szerkezete, míg az npt.cpt a futás folytatásához szükséges állapotot tartalmazza. Az NPT szakasz után érdemes ellenőrizni, hogy a rendszer sűrűsége és nyomása ésszerűen stabilizálódott-e.
A következő paranccsal kiírattatható a nyomás változása egy pressure.xvg fájlba:
gmx energy -f nvt.edr -o pressure.xvg
A gmx energy futtatásakor a promptnál a 17 0 értékeket kell megadni: a 17 a rendszer Pressure tagjának kiválasztását jelenti, a 0 pedig a bevitelt zárja le.
Melyet később akár egy rövid python script (pressure_plot.py) segítségével ki is lehet rajzoltatni.
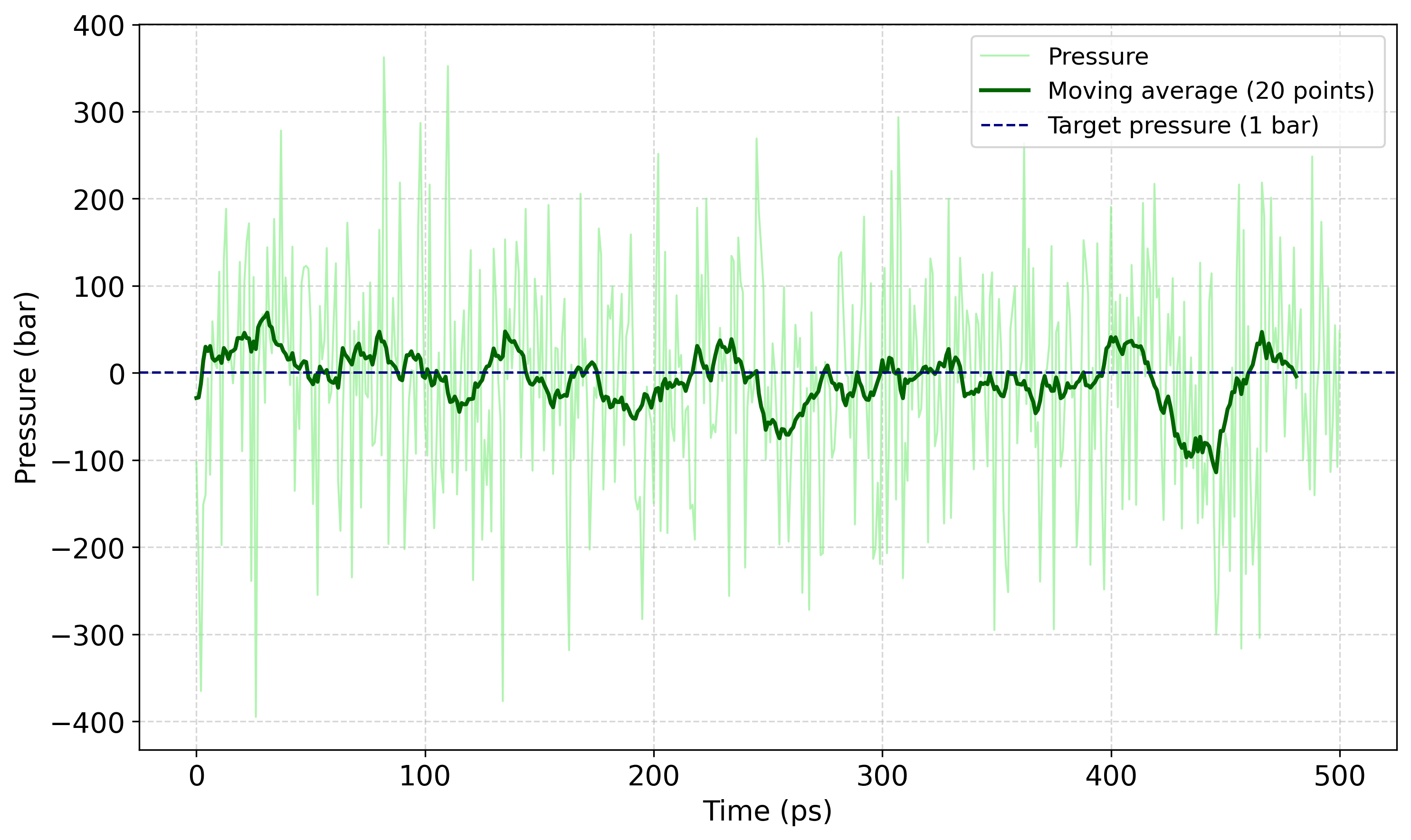
Mintavételezési szakasz
A két kiegyenlítési szakasz után a rendszer már megfelelően stabilizált a kívánt hőmérsékleten és nyomáson, ezért a pozíciós restrikciók feloldhatók, és megkezdhető a mintavételezési molekuladinamikai futás. A mintavételezési szakasz futtatáshoz szükség van egy md.mdp fájlra is, amely a hosszabb, restrikciómentes molekuladinamikai szimuláció paramétereit tartalmazza.
Minta md.mdp fájl:
title = OPLS Lysozyme MD run
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 5000000 ; 2 * 2500000 = 10000 ps (10 ns)
dt = 0.002 ; 2 fs
; Output control
nstxout = 0 ; suppress bulky .trr file by specifying
nstvout = 0 ; 0 for output frequency of nstxout,
nstfout = 0 ; nstvout, and nstfout
nstenergy = 5000 ; save energies every 10.0 ps
nstlog = 5000 ; update log file every 10.0 ps
nstxout-compressed = 5000 ; save compressed coordinates every 10.0 ps
compressed-x-grps = System ; save the whole system
; Bond parameters
continuation = yes ; Restarting after NPT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
; vdW
rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
rvdw-switch = 1.0
vdw-modifier = force-switch
DispCorr = No
; Electrostatics
rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm)
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = System
tau_t = 1.0
ref_t = 298
; Pressure coupling is on
pcoupl = C-rescale
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 5.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = no ; Velocity generation is off
A futtatási bemenet (md.tpr) előkészítése:
gmx grompp -f inputs/md.mdp -c npt.gro -t npt.cpt -p topol.top -o md.tpr
Ebben a parancsban a -c npt.gro az NPT kiegyenlítés végső szerkezetét, a -t npt.cpt pedig a folytatáshoz szükséges checkpoint fájlt adja meg.
A produkciós szimuláció indítása:
gmx mdrun -deffnm md
A futás során a GROMACS létrehozza a mintavételezési szimulációhoz tartozó szokásos kimeneti fájlokat, például a logfájlt, az energiafájlt, a végső koordinátafájlt és a checkpoint állományt. A md alapnév alapján ezek a fájlok ennek megfelelő előtaggal jönnek létre. A GROMACS dokumentáció szerint az mdrun minden ilyen szakaszban a .tpr bemenet alapján végzi el a megadott feladatot, és írja a hozzá tartozó md.log, md.edr, md.gro és md.cpt fájlokat. A trajektóriát a md.xtc tartalmazza.
Analízis: backbone RMSD számolás
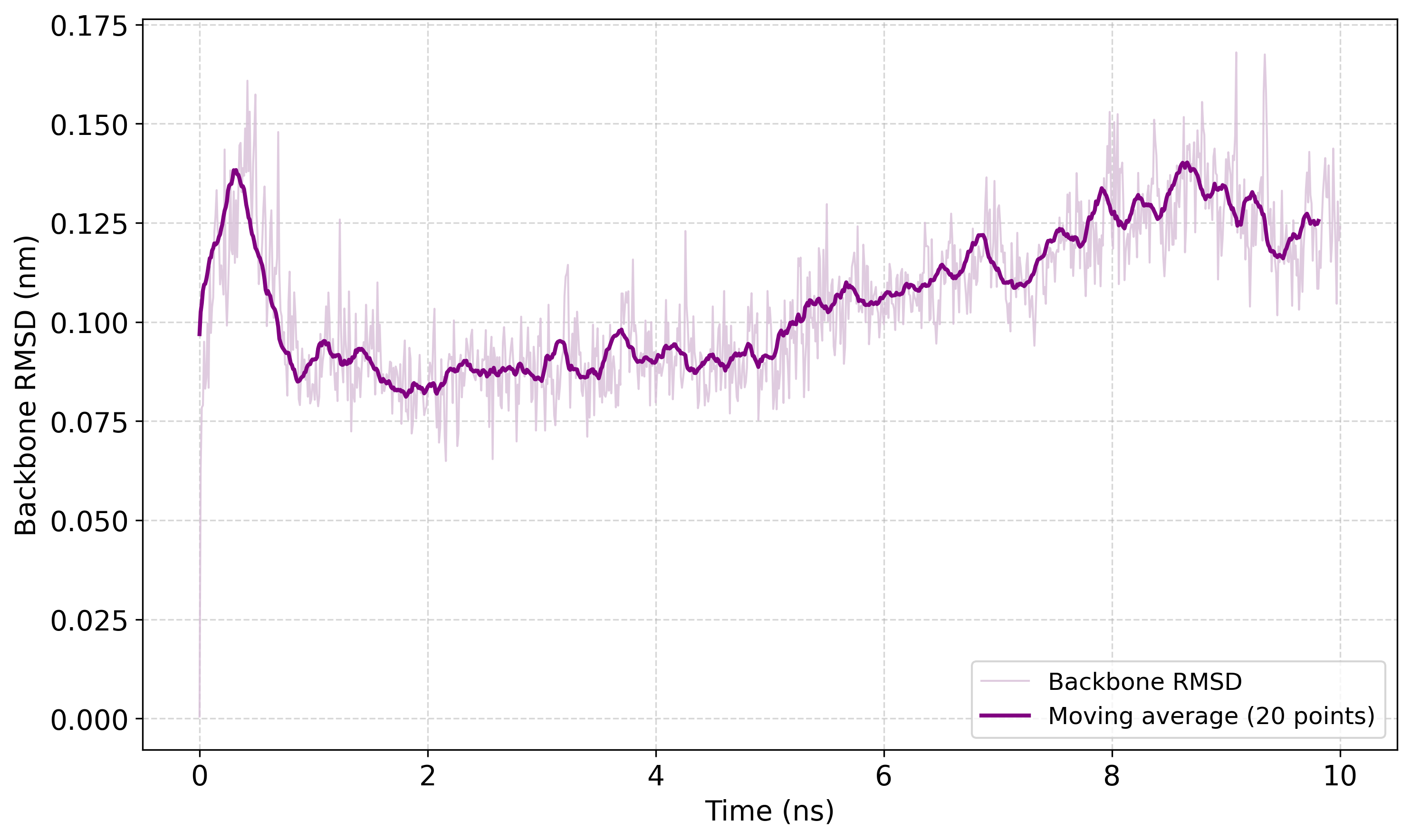
A mintavételezési futás után az egyik legegyszerűbb és leggyakrabban használt szerkezeti analízis a backbone RMSD meghatározása. Az RMSD számolás előtt célszerű a trajektóriát úgy utófeldolgozni, hogy a periódusos határfeltételek ne torzítsák az eredményt. Ennek oka, hogy a fehérje a szimuláció során diffundálhat a dobozban, és emiatt a trajektórián „szétszakadtnak” vagy elugrónak tűnhet.
A periódicitás korrigálásához a gmx trjconv parancs használható:
gmx trjconv -s md.tpr -f md.xtc -o md_PBC.xtc -pbc mol -center
A parancs futtatásakor a tutorial szerint először az 1 számú Protein csoportot kell kiválasztani a középre helyezéshez, majd a 0 számú System csoportot a teljes rendszer kiírásához. Az így előállított md_PBC.xtc trajektórián már megszűnnek a periodicitásból eredő zavaró vizuális és geometriai hatások, ezért a további analíziseket ezen a korrigált trajektórián érdemes elvégezni.
A backbone RMSD számításához a gmx rms parancs használható:
gmx rms -s md_0_10.tpr -f md_0_10_noPBC.xtc -o rmsd.xvg -tu ns
A parancs futtatásakor a tutorial alapján mind az illesztéshez (least-squares fit), mind magához az RMSD számításhoz a 4 számú Backbone csoportot kell kiválasztani. A -tu ns opció azt adja meg, hogy az időtengely nanoszekundumban jelenjen meg, ami hosszabb trajektóriák esetén áttekinthetőbb, mint a pikoszekundumos skála.
Az így kapott rmsd.xvg fájl az idő függvényében tartalmazza a backbone RMSD értékeket. A lysozyme tutorial szerint ez a görbe azt mutatja meg, hogy a fehérje gerince mennyire tér el a referenciaszerkezettől a szimuláció során. A példában a referencia a minimizált és kiegyenlített rendszer szerkezete, mert a md_0_10.tpr ezt reprezentálja.
Az rmsd.xyv-t később akár egy rövid python script (rmsd_plot.py) segítségével ki is lehet rajzoltatni.